L’anemia falciforme o drepanocitosi è una patologia rara a carico dell’emoglobina, la proteina che trasporta l’ossigeno nel sangue. L’anomalia genetica alla base di questa malattia, molto frequente in Italia, determina un danno ai globuli rossi, con conseguenze invalidanti per i pazienti. Negli ultimi anni sono stati fatti notevoli passi avanti nel trattamento e abbiamo a disposizione nuove armi per questi pazienti. Vediamo quali.
Come si manifesta la malattia?
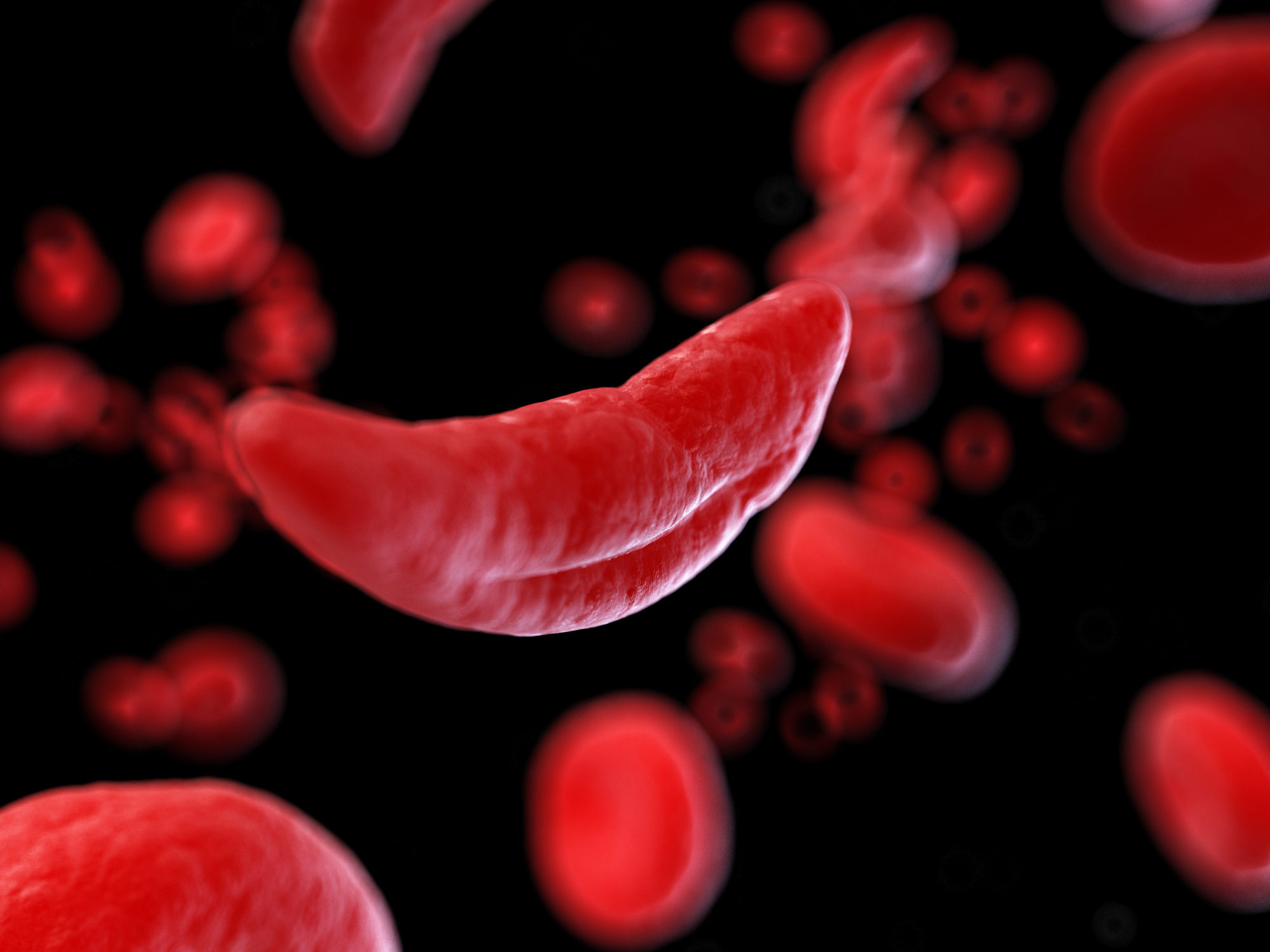
L’anemia falciforme è una malattia causata da mutazioni del gene dell’emoglobina, che determinano la formazione di una proteina alterata, l’emoglobina S, dall’inglese sickle (falce), la tipica forma assunta dai globuli rossi di questi pazienti.
Viene trasmessa da genitori portatori del gene difettoso con modalità autosomica recessiva: in questo caso, per manifestare la malattia è necessario ereditare due copie anormali del gene, uno da ciascun genitore.
Nei soggetti affetti, l’emoglobina S è preponderante sulle emoglobine normali e precipita formando dei filamenti che modificano la forma dei globuli rossi rendendoli rigidi, con conseguente ridotta capacità di trasportare l’ossigeno nell’organismo e di attraversare i piccoli capillari. I globuli rossi a falce vengono eliminati più velocemente dalla milza e possono formare ostruzioni dei vasi sanguigni con conseguente danno ai tessuti che si manifestano con crisi dolorose, innescate in particolare in condizioni di ridotta disponibilità di ossigeno, inclusi gli sforzi fisici. Inoltre, l’ostruzione del flusso ematico può causare nel tempo danni irreversibili a diversi organi, tra cui milza, reni, cervello e ossa, con impatto sull’aspettativa di vita.
Oltre alle crisi, che si manifestano con frequenza e gravità variabile, i pazienti presentano i sintomi tipici di un’anemia, cioè stanchezza, vertigini, pallore, mancanza di respiro e aumentata frequenza cardiaca. I soggetti affetti sono anche maggiormente sensibili alle infezioni.
Quali sono le armi a disposizione per il trattamento di questi pazienti?
In primo luogo, è necessario trattare l’anemia e gestire le crisi, per ridurre il dolore e limitare le complicanze. In caso di grave anemia può essere necessario sottoporre il paziente ad una trasfusione di sangue. Durante una crisi, il paziente deve essere adeguatamente idratato e può assumere antidolorifici per il controllo del dolore.
Per la prevenzione delle infezioni i pazienti devono essere sottoposti ad adeguata vaccinazione, inclusa la somministrazione del vaccino contro le infezioni da pneumococco e dei vaccini antinfluenzali.
Per la prevenzione delle crisi è possibile usare l’idrossiurea: si tratta di un farmaco citotossico, utilizzato in diverse malattie del sangue, che nei pazienti con anemia falciforme aumenta la concentrazione di emoglobina fetale, interferendo con la precipitazione della emoglobina S che danneggia i globuli rossi. Tuttavia, il farmaco non è sempre efficace.
Nei casi più gravi, il trapianto di cellule staminali ematopoietiche rappresenta l’unica alternativa potenzialmente curativa. Non è, però, semplice trovare un donatore compatibile e la procedura presenta numerosi effetti indesiderati, anche molto gravi, oltre al rischio di rigetto.
Quali sono le prospettive future?
Considerata la mancanza di terapie in grado di curare la malattia e prevenire i danni a lungo termine, numerosi studi sono stati condotti per trovare possibili alternative.
La prima terapia mirata approvata in Europa è rappresentata da un anticorpo monoclonale, il crizanlizumab, che impedisce l’adesione cellulare, prevenendo le crisi ricorrenti.
Voxelotor è, invece, un farmaco somministrato per via orale, da solo o in associazione con l’idrossiurea, in un’unica somministrazione giornaliera, in grado di contrastare la precipitazione dell’emoglobina S e il conseguente danno ai globuli rossi. È stato approvato nel mese di dicembre 2021 dall’Agenzia Europea dei Medicinali (EMA) per i pazienti affetti da anemia a cellule falciformi con età ³12 anni.
Infine, è in fase avanzata di studio la terapia genica: l’obiettivo di questa terapia è quello di consentire la sintesi di un’emoglobina normale inserendo il gene sano nelle cellule del paziente, senza dover ricorrere al trapianto da donatore compatibile. In questo caso, le cellule del singolo paziente vengono prelevate, modificate geneticamente tramite un vettore virale e poi reinfuse nel paziente stesso. Inoltre, è in studio anche la terapia genica che utilizza l’editing genomico con la tecnologia innovativa CRISPR-Cas9.
Referenze
- Meisel R. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N Engl J Med. 2021 Jun 10;384(23):e91. doi: 10.1056/NEJMc2103481. PMID: 34107195.
- Howard J, Ataga KI, Brown RC, Achebe M, Nduba V, El-Beshlawy A, Hassab H, Agodoa I, Tonda M, Gray S, Lehrer-Graiwer J, Vichinsky E. Voxelotor in adolescents and adults with sickle cell disease (HOPE): long-term follow-up results of an international, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Haematol. 2021 May;8(5):e323-e333. doi: 10.1016/S2352-3026(21)00059-4. Epub 2021 Apr 7. PMID: 33838113.
- https://www.ema.europa.eu/en/news/new-treatment-sickle-cell-disease
- https://www.ema.europa.eu/en/medicines/human/EPAR/adakveo